About the science
Alzheimer's disease (AD) and other neurodegenerative diseases as lysosomal protein storage diseases
The true underlying cause of AD has eluded science to date, but substantial learnings about pathophysiologic processes and the role of amyloid accumulation in some cases has increased our understanding. Treating the underlying cause of AD and other diseases will be critical to making ultimate progress in this health care crisis.
Amyloid beta (Aβ) accumulation is a cause of AD. Since the hypothesis was introduced more than 30 years ago, evidence supporting accumulation of Aβ in the brain as a cause of AD has grown (Selkoe and Hardy, 2016). One of the most compelling facts supporting this hypothesis is that individuals with Down syndrome, who likely have increased production of the amyloid precursor protein (APP) due to an extra copy of the APP gene on the extra chromosome 21, are predisposed to developing early-onset AD (Sirisi et al., 2024; Fortea et al., 2021; Selkoe and Hardy, 2016). The extra production of APP results in more toxic Aβ being formed; however, the Aβ degradation rate is not increased, which leads to accumulation of excess Aβ. In individuals with AD, accumulation of Aβ aggregates is consistently found in the brain, and Aβ in the cerebrospinal fluid (CSF) is used as a biomarker of disease. Studies have shown that low CSF Aβ42, which is considered the most toxic Aβ peptide isoform (Annunziata et al., 2013), and amyloid positivity on positron emission tomography (PET) imaging precede other manifestations of AD by years. In animal models, soluble Aβ oligomers decrease the number of synapses, inhibit long-term potentiation, and induce tau hyperphosphorylation.
Current management of AD. There is no cure for AD. Monoclonal antibodies that bind and neutralize amyloid were the first therapies to target the underlying biology of AD. The success of anti-Aβ monoclonal antibodies validates Aβ accumulation as an important target for treatment (Sirisi et al., 2024). However, a significant unmet medical need remains for a treatment that can stop or reverse AD pathological processes.
Lysosomal dysfunction is increasingly recognized as a contributor to amyloid pathology in AD and other neurodegenerative diseases. Multiple lysosomal storage disorders have demonstrated amyloid accumulation in patient brains and animal models (Table 1). For example, Gaucher disease is associated with α-synuclein aggregation, Sanfilippo syndrome shows signs of tau pathology, and sialidosis has been linked to Aβ accumulation (Annunziata et al., 2013; Ohmi et al., 2009). These findings suggest that impaired lysosomal clearance mechanisms may broadly contribute to protein aggregation across distinct neurodegenerative contexts.
There is growing evidence that lysosomal dysfunction and oligomerization in the lysosomal compartment have a key role in Aβ accumulation and tissue injury in AD (Cha et al., 2025; Chou et al., 2025; Tian et al., 2025; Bretou et al., 2024; Jacquet et al., 2024; Askenazi et al., 2023; Brase et al., 2023; Im et al., 2023; Chen et al., 2020; Xiao et al., 2015). In the dysfunctional lysosome, Aβ proteins are inadequately degraded due to a relative deficiency of proteases, which leads to lysosomal protein accumulation followed by aggregation to form oligomers and fibrils. The intracellular fibril formation then causes lysosomal rupture leading to neurodegeneration, accumulates into extracellular plaque causing inflammation, and activates multiple other downstream processes.
Upstream determinants of AD pathology have been directly linked to lysosomal dysfunction and create a harmful feedback loop (Im et al., 2023; Annunziata et al., 2013). C-terminal fragments of APP, linked to early onset AD in Down syndrome, disrupt lysosomal function, which promotes the accumulation of Aβ. And this relationship is causal – reducing C-terminal fragments of APP restores lysosomal activity and mitigates Aβ accumulation.
Protective protein/cathepsin A (PPCA) can catalytically degrade Aβ and thereby reduce both intracellular and extracellular accumulating amyloid. PPCA is a potent carboxypeptidase enzyme that can processively degrade the C-terminus of Aβ42, the essential structure required for aggregation of Aβ42 and oligomer stability, leading to degradation of Aβ even within an oligomeric form. This catalytic degradation means that PPCA can degrade potentially thousands of amyloid monomers and oligomers as compared with only a few for the stoichiometric monoclonal antibody approach. In addition to its direct proteolytic effect, PPCA binds and enhances the transport of neuraminidase 1 (NEU1) to the lysosome leading to 2-fold higher levels of the enzyme, which has an important impact on processing of lysosomes and delaying lysosomal exocytosis to the exterior. This additional enhancement of NEU1 activity likely further enhances the degradative power of PPCA and the degradation of APP, which is a sialylated protein.
Enhancing Aβ degradation has the potential to reduce oligomers and fibril formation in neurons and, by clearing intracellular accumulation, to reduce dysfunction and rupture in neurons. Additionally, PPCA enzyme supplementation has the potential to enhance microglia degradation of extracellular amyloid during exophagy (Jacquet et al., 2024). Enhancing PPCA activity across multiple cell types may mitigate AD pathology by improving lysosomal function and promoting clearance of pathogenic protein aggregates.
Treating AD models with increased PPCA via gene therapy can reduce or prevent amyloid plaque. We have studied the idea that the accumulation of amyloids both extracellularly and intracellularly is due to a relative deficiency of proteases needed to cleave the very difficult to digest oily protein oligomers and fibrils that accumulate with age, particularly within the lysosomal compartment. Work by Dr. Alessandra d’Azzo at St. Jude Children’s Research Hospital in Memphis and our own research team have demonstrated that PPCA can degrade Aβ in oligomeric and higher molecular weight aggregates as well as prevent Aβ aggregation in vitro. Other lysosomal proteases and cathepsins such as Cathepsin L do not have this activity. The fact that PPCA can cleave multiple higher order amyloid forms opens the door to a more complete reversal of Aβ accumulation intraneuronally and a more complete reduction of plaque and extracellular amyloid. By delivering supplemental PPCA to the brain using AAV gene therapy in murine models of AD, we have also shown that the increased PPCA enzyme levels in the lysosomes of neurons reduce amyloid plaque accumulation outside the cell more effectively than monoclonal antibodies targeting Aβ42. This is because clearing the amyloid and toxic oligomers contained within the neuronal cell lysosomal compartment reduces the rupture of neurons and exocytosis of undigested amyloid outside the cell where it can become plaque.
This insight is a fundamental change in understanding of how and why amyloids accumulate and cause disease and opens the door to a profound single-shot treatment to reduce or prevent AD.
| Lysosomal Disease (Gene) | Type of Amyloid Deposition | Observation |
|---|---|---|
| Batten Disease/CLN2 (TPP1) | Amyloid like proteo-lipid material | Extracellular Aβ-amyloid accumulation and neuronal cell death in patient brain |
| Gaucher disease (GBA1) | α-synuclein | α-synuclein accumulates in Lewy bodies and neurites in brain & spinal cord of patients leading to Parkinsonian symptoms |
| MPS IIIA & MPS IIIB (SGSH & NAGLU) | α-synuclein Phospho-Tau Increased Aβ-amyloid |
Amyloid protein aggregates detected in patient postmortem brains. In addition, aggregation of amyloid proteins in neuronal cell bodies in mouse model brain |
| Sialidosis (Neu1) | Aβ-amyloid | Extracellular amyloid accumulation in Neu1-/- mouse brain |
Delivery of PPCA enhances lysosomal function and degradation of amyloid
Protective protein cathepsin A (PPCA) has two distinct functions, both of which serve to break down amyloid aggregates:
-
- In the endosomal/lysosomal compartment, PPCA has carboxypeptidase activity at acidic pH directed towards substrates with hydrophobic amino acids.
- PPCA stabilizes neuraminidase 1 and beta-galactosidase to form a multi-enzyme complex, actively enhancing lysosomal clearance of proteins.
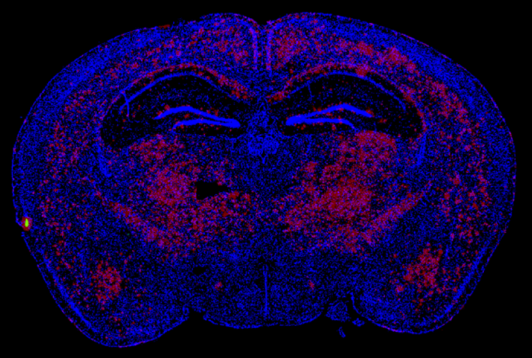
AAV9 PPCA gene therapy in murine model of severe AD
Vehicle treated
PPCA treated
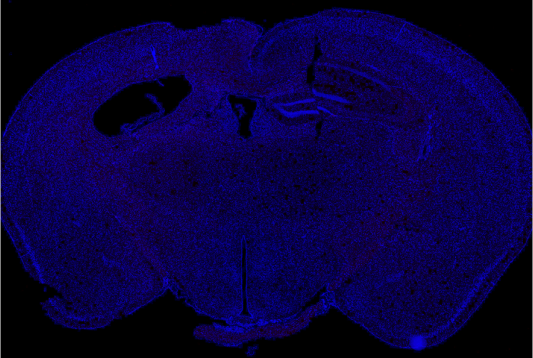
12 weeks following AAV delivery of PPCA we see near complete reversal in an aggressive murine model of severe AD (5xFAD), with the extent of clearance correlating with PPCA brain exposure. (Red=Amyloid)
Vehicle treated
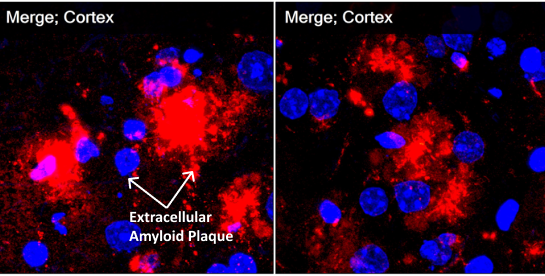
PPCA treated
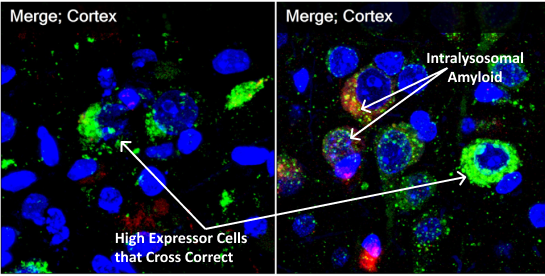
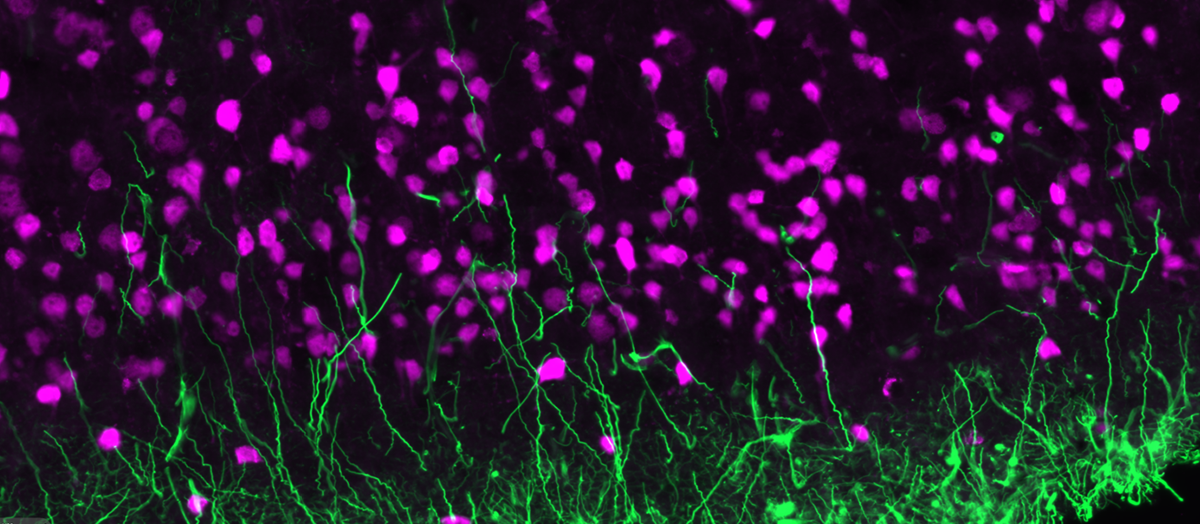
Immunohistochemistry staining shows massive fluffy red-staining amyloid in control vehicle-treated murine models outside cells but also in the perinuclear lysosomes. On the right in the treated animals, green-staining PPCA is in the perinuclear lysosomes with tiny pieces of red amyloid (now showing yellow in the merged pictures) colocalizing with PPCA. These pictures show that lysosomal PPCA can be cross-corrected from high expression cells, increase PPCA in many neurons and enhance lysosomal degradation of amyloid. (Red=Amyloid; Green=PPCA)
References
- Annunziata I, Patterson A, Helton D, et al. (2013) Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-beta secretion via deregulated lysosomal exocytosis. Nat Commun 4: 2734. DOI: 10.1038/ncomms3734.
- Askenazi M, Kavanagh T, Pires G, et al. (2023) Compilation of reported protein changes in the brain in Alzheimer’s disease. Nat Commun 14(1): 4466. DOI: 10.1038/s41467-023-40208-x.
- Brase L, You SF, D’Oliveira Albanus R, et al. (2023) Single-nucleus RNA-sequencing of autosomal dominant Alzheimer disease and risk variant carriers. Nat Commun 14(1): 2314. DOI: 10.1038/s41467-023-37437-5.
- Bretou M, Sannerud R, Escamilla-Ayala A, et al. (2024) Accumulation of APP C-terminal fragments causes endolysosomal dysfunction through the dysregulation of late endosome to lysosome-ER contact sites. Dev Cell 59(12): 1571–1592.e9. DOI: 10.1016/j.devcel.2024.03.030.
- Cha YJ, Kim YK, Lim YJ, et al. (2025) Lipidomic Network Analysis Reveals Amyloid-beta-Induced Lysosomal Lipid Accumulation in the Cortex and Hippocampus of 5xFAD Mice. J Proteome Res 24(7): 3389–3398. DOI: 10.1021/acs.jproteome.4c01133.
- Chen WT, Lu A, Craessaerts K, et al. (2020) Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 182(4): 976–991.e19. DOI: 10.1016/j.cell.2020.06.038.
- Chou CC, Vest R, Prado MA, et al. (2025) Proteostasis and lysosomal repair deficits in transdifferentiated neurons of Alzheimer’s disease. Nat Cell Biol 27(4): 619–632. DOI: 10.1038/s41556-025-01623-y.
- Fortea J, Zaman SH, Hartley S, et al. (2021) Alzheimer’s disease associated with Down syndrome: a genetic form of dementia. Lancet Neurol 20(11): 930–942. DOI: 10.1016/S1474-4422(21)00245-3.
- Im E, Jiang Y, Stavrides PH, et al. (2023) Lysosomal dysfunction in Down syndrome and Alzheimer mouse models is caused by v-ATPase inhibition by Tyr(682)-phosphorylated APP betaCTF. Sci Adv 9(30): eadg1925. DOI: 10.1126/sciadv.adg1925.
- Jacquet RG, Gonzalez Ibanez F, Picard K, et al. (2024) Microglia degrade Alzheimer’s amyloid-beta deposits extracellularly via digestive exophagy. Cell Rep 43(12): 115052. DOI: 10.1016/j.celrep.2024.115052.
- Ohmi K, Kudo LC, Ryazantsev S, et al. (2009) Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc Natl Acad Sci U S A 106(20): 8332–8337. DOI: 10.1073/pnas.0903223106.
- Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8(6): 595–608. DOI: 10.15252/emmm.201606210.
- Sirisi S, Sanchez-Aced E, Belbin O, et al. (2024) APP dyshomeostasis in the pathogenesis of Alzheimer’s disease: implications for current drug targets. Alzheimers Res Ther 16(1): 144. DOI: 10.1186/s13195-024-01504-w.
- Tian Q, Li J, Wu B, et al. (2025) APP lysine 612 lactylation ameliorates amyloid pathology and memory decline in Alzheimer’s disease. J Clin Invest 135(1). DOI: 10.1172/JCI184656.
- Xiao Q, Yan P, Ma X, et al. (2015) Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing Abeta Generation and Amyloid Plaque Pathogenesis. J Neurosci 35(35): 12137–12151. DOI: 10.1523/JNEUROSCI.0705-15.2015.